OSTEOCLASTS IN BONE DISEASE
 The skeleton is composed of three cell types that work together to form and maintain the bone structure: bone forming osteoblasts, bone resorbing osteoclasts, and mechanosensing osteocytes. Under conditions of homeostasis, bone resorption by osteoclasts is balanced by bone formation by osteoblasts.
The skeleton is composed of three cell types that work together to form and maintain the bone structure: bone forming osteoblasts, bone resorbing osteoclasts, and mechanosensing osteocytes. Under conditions of homeostasis, bone resorption by osteoclasts is balanced by bone formation by osteoblasts.
Dysregulation of osteoclastic bone resorption contributes to the pathologies observed in many diseases, including osteopetrosis and fibrous dysplasia. Our laboratory focuses on the role of osteoclast in these pathologies and the relationship between osteoclast resorption and formation by osteoblasts, termed coupling. Through coupling, osteoclastic bone resorption contributes to the formation of fragile bone with an inherent risk of fracture. Our research investigates the mechanistic pathways which control osteoclastogenesis to identify potential therapeutic targets to prevent the formation of fragile bone and improve the quality of bone in these diseases.

Our group has found that bone marrow is replaced by osteoblastic precursors in multiple models of osteoclast rich osteopetrosis. This osteoblastic cell expansion is osteoclast-dependent and we hypothesize that this is a disease of aberrant coupling. We are currently investigating the mechanism by which osteoclasts promote osteoblastic cell accumulation using transcriptional analysis of mutant osteoclasts from mouse and zebrafish models of osteoclast rich osteopetrosis to identify putative coupling factors.

Autosomal dominant osteopetrosis type II is due to mutations in CLCN7, encoding a chloride channel essential for normal acidification of the osteoclasts resorption lacunae. Bone resorption is impaired. Despite this decreased bone resorption activity, bone formation continues and overfills areas of resorption. Using a variety of mouse models of ADOII, we are investigating whether interactions between mutant osteoclasts and osteoblast at the site of resorption drive this phenomenon of overflow bone remodeling. This project is a collaboration with Dr. Thomas Anderson, University of Southern Denmark (Andersen Lab website).

Fibrous dysplasia (FD) is caused by somatic activating mutations in GNAS and affects approximately 1 in 5000 individuals. Activation of GNAS causes focal bone lesions that result in pain, deformity and fracture susceptibility. FD lesions are mosaics of wildtype and mutant cells and are characterized by an accumulation of immature osteoblast lineage cells as well as robust osteoclastogenesis with increased bone turnover. Receptor activator of NFkB ligand (RANKL), the key cytokine driving osteoclast differentiation, is highly expressed by osteoblastic cells in FD lesions and circulating RANKL is elevated in people with FD. Denosumab, a neutralizing antibody against RANKL, prevents osteoclast formation and decreases lesion size in FD. However, denosumab can inhibit skeletal growth and withdrawal can cause rebound bone resorption and fractures. Thus, alternative therapies are needed. If we could understand how anti-RANKL inhibits FD bone lesions, we might be able to discover new targets or ways of improving denosumab therapy. Our work uses an inducible model of FD lesions developed by the Yingzi Yang lab to try to dissect the cellular target of RANKL inhibition in FD lesions. This project was started with pilot funding from the FD/MAS alliance.
GUT MICROBIOME-BONE AXIS
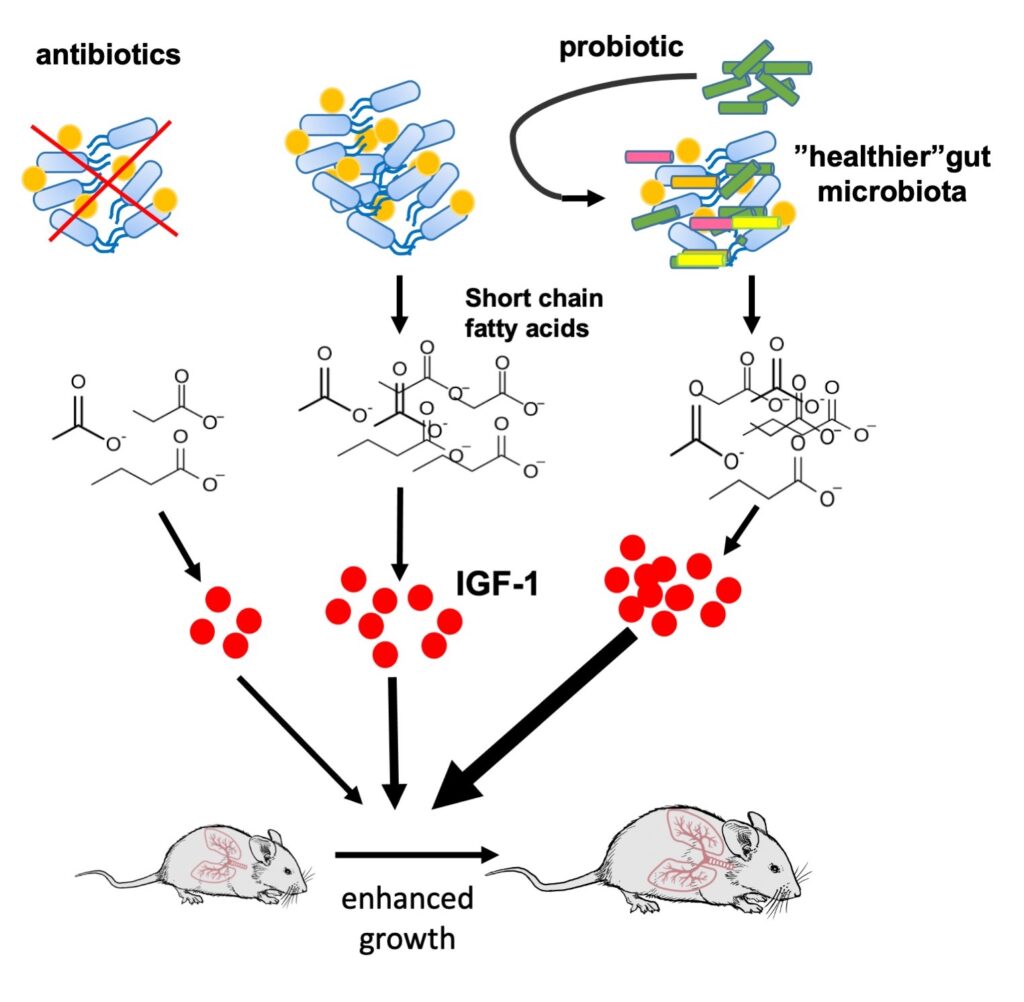 The gut harbors a complex ecosystem of microorganisms, collectively referred to as to the gut microbiota. The initial colonization of gut microbiota is generally thought to be established at birth, developing toward a stable adult-like microbial community structure by age 3 years. We previously demonstrated an important role of gut microbiota in modulating serum IGF-1 to impact bone homeostasis. We have shown that a colonization of germ-free mice induces IGF-1 and promotes bone formation and growth. Currently, we are studying the mechanisms by which the GM modulates serum IGF-1. We are interested in the mechanisms by which gut microbiota regulates IGF-1 and bone formation in healthy conditions and pathologic bone loss conditions such as aging, malnutrition, immune dysfunction and cystic fibrosis. We use genetic mutant models, germ-free mice, fecal transplantation, in vivo dietary intervention and antibiotic treatment to investigate this question.
The gut harbors a complex ecosystem of microorganisms, collectively referred to as to the gut microbiota. The initial colonization of gut microbiota is generally thought to be established at birth, developing toward a stable adult-like microbial community structure by age 3 years. We previously demonstrated an important role of gut microbiota in modulating serum IGF-1 to impact bone homeostasis. We have shown that a colonization of germ-free mice induces IGF-1 and promotes bone formation and growth. Currently, we are studying the mechanisms by which the GM modulates serum IGF-1. We are interested in the mechanisms by which gut microbiota regulates IGF-1 and bone formation in healthy conditions and pathologic bone loss conditions such as aging, malnutrition, immune dysfunction and cystic fibrosis. We use genetic mutant models, germ-free mice, fecal transplantation, in vivo dietary intervention and antibiotic treatment to investigate this question.